Introduction
This vignette is a workflow template including import, conversion to a standard format and highlighting inter-software proteinGroup denotation differences with flowTraceR.
Import
Import your data
Importing the output files from each software can be easily performed with data.table::fread().
diann <- data.table::fread("DIRECTORY/dia-nn_file.tsv")
spectronaut <- data.table::fread("DIRECTORY/spectronaut_file.tsv")
mq_evidence <- data.table::fread("DIRECTORY/maxquant_evidence.txt")
mq_proteinGroups <- data.table::fread("DIRECTORY/maxquant_proteinGroups.txt")
pd_psm <- data.table::fread("DIRECTORY/pd_PSMs.txt")Examples
Some examples are provided to explore the workflow.
#DIA-NN
diann <- flowTraceR::get_example("DIA-NN")
#Spectronaut
spectronaut <- flowTraceR::get_example("Spectronaut")
#MaxQuant
mq_evidence <- flowTraceR::get_example("MaxQuant")[["evidence"]]
mq_proteinGroups <- flowTraceR::get_example("MaxQuant")[["proteinGroups"]]
#PD
pd_psm <- flowTraceR::get_example("PD")Conversion to standardized format
The input data can be converted to a standardized output format on precursor, modified peptide and proteingroup level. The generated columns with flowTraceR are appended to the submitted data without any filtering performed. The generated columns are denoted with the prefix traceR. Note that only the modifications UniMod:35 (Oxidation) and UniMod:4 (Carbamidomethyl) are supported by flowTraceR. A column with the appendix unknownMods is generated to potentially filter modifications which are not supported: if TRUE, an unknown modification is detected.
Precursor
For converting the precursor level use convert_precursor().
diann_precursor_converted <- convert_precursor(input_df = diann, software = "DIA-NN")
spectronaut_precursor_converted <- convert_precursor(input_df = spectronaut, software = "Spectronaut")
mq_precursor_converted <- convert_precursor(input_df = mq_evidence, software = "MaxQuant")
pd_precursor_converted <- convert_precursor(input_df = pd_psm, software = "PD")Modified Peptides
For converting the modified peptide level use convert_modified_peptides().
diann_peptides_converted <- convert_modified_peptides(input_df = diann, software = "DIA-NN")
spectronaut_peptides_converted <- convert_modified_peptides(input_df = spectronaut, software = "Spectronaut")
mq_peptides_converted <- convert_modified_peptides(input_df = mq_evidence, software = "MaxQuant")
pd_peptides_converted <- convert_modified_peptides(input_df = pd_psm, software = "PD")ProteinGroups
For converting the proteinGroup level use convert_proteingroups().
diann_proteinGroups_converted <- convert_proteingroups(input_df = diann, software = "DIA-NN")
spectronaut_proteinGroups_converted <- convert_proteingroups(input_df = spectronaut, software = "Spectronaut")
mq_proteinGroups_converted <- convert_proteingroups(input_df = mq_proteinGroups, software = "MaxQuant")
pd_proteinGroups_converted <- convert_proteingroups(input_df = pd_psm, software = "PD")All Levels
For converting precursor, modified peptide and proteingroup level at once use convert_all_levels().
diann_all_converted <- convert_all_levels(input_df = diann, software = "DIA-NN")
spectronaut_all_converted <- convert_all_levels(input_df = spectronaut, software = "Spectronaut")
mq_all_converted <- convert_all_levels(input_df = mq_evidence, input_MQ_pg = mq_proteinGroups, software = "MaxQuant")
pd_all_converted <- convert_all_levels(input_df = pd_psm, software = "PD")Analyzing Conversion
Since only the modifications UniMod:35 (Oxidation) and UniMod:4 (Carbamidomethyl) are currently supported, flowTraceR provides functions to analyze the conversion and shows how much unknown modifications are present in the dataset with analyze_unknown_mods().
#For one software example - equivalent for others.
#Proteome Discoverer
#Reports
pd_precursor_report_unknown_mods <- analyze_unknown_mods(input_df = pd_precursor_converted, level = "precursor", plot = FALSE)
pd_peptides_report_unknown_mods <- analyze_unknown_mods(input_df = pd_peptides_converted, level = "modified_peptides", plot = FALSE)
#Plots
pd_precursor_plot_unknown_mods <- analyze_unknown_mods(input_df = pd_precursor_converted, level = "precursor", plot = TRUE, plot_characteristic = "absolute")
pd_peptides_plot_unknown_mods <- analyze_unknown_mods(input_df = pd_peptides_converted, level = "modified_peptides", plot = TRUE, plot_characteristic = "relative")Example precursor level
kableExtra::kable(pd_precursor_report_unknown_mods)| Unknown_Modifications | absolute_count | relative_count |
|---|---|---|
| FALSE | 8 | 88.9 |
| TRUE | 1 | 11.1 |
pd_precursor_plot_unknown_mods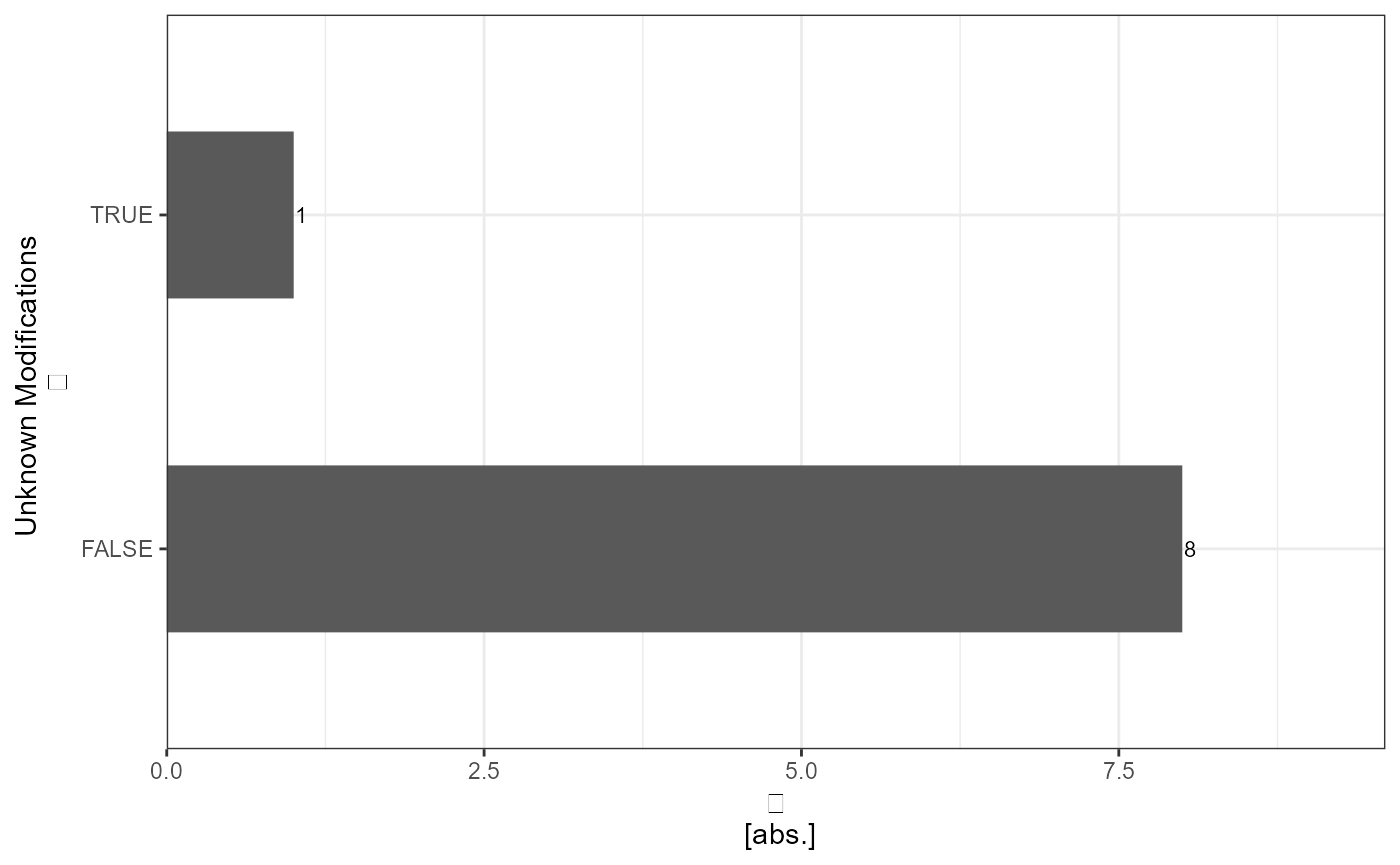
Tracing inter-software differences
For binary software comparisons flowTraceR allows to trace inter-software differences based on the standardized flowTraceR format. Each identification is classified as common - identified in both analyses or as unique - specific to one analysis.
For each individual level
#Binary Comparison - DIA-NN vs. Spectronaut
#ProteinGroup level
traced_proteinGroups <- trace_level(input_df1 = diann_all_converted , input_df2 = spectronaut_all_converted, analysis_name1 = "DIA-NN", analysis_name2 = "Spectronaut", level = "proteinGroups", filter_unknown_mods = TRUE)
#Peptide level
traced_peptides <- trace_level(input_df1 = diann_all_converted, input_df2 = spectronaut_all_converted, analysis_name1 = "DIA-NN", analysis_name2 = "Spectronaut", level = "modified_peptides", filter_unknown_mods = TRUE)
#Precursor level
traced_precursor <- trace_level(input_df1 = diann_all_converted, input_df2 = spectronaut_all_converted, analysis_name1 = "DIA-NN", analysis_name2 = "Spectronaut", level = "precursor", filter_unknown_mods = TRUE)All levels
#Binary Comparison - DIA-NN vs. Spectronaut
#trace all levels in one step
traced_all <- trace_all_levels(input_df1 = diann_all_converted, input_df2 = spectronaut_all_converted, analysis_name1 = "DIA-NN", analysis_name2 = "Spectronaut", filter_unknown_mods = TRUE)Connect traced levels
Combine two levels after categorization in unique and common entries. Possible connections are proteinGroup or modified peptide with precursor categorization.
#ProteinGroup level
DIANN_connected_proteinGroup <- connect_traceR_levels(input_df = traced_all[["DIA-NN"]], level = "proteinGroups")
Spectronaut_connected_proteinGroup <- connect_traceR_levels(input_df = traced_all[["Spectronaut"]], level = "proteinGroups")
#Peptide level
DIANN_connected_peptides <- connect_traceR_levels(input_df = traced_all[["DIA-NN"]], level = "modified_peptides")
Spectronaut_connected_peptides <- connect_traceR_levels(input_df = traced_all[["Spectronaut"]], level = "modified_peptides")Show software differences for modified peptide/proteinGroup denotations
Generate a report or visualize the output of connecting the flowTraceR levels on proteinGroup_precursor or modified.peptides_precursor categorization in:
- common_common
- common_unique
- unique_common
- unique_unique
#Example for proteinGroup level
#*Plots*
#upper level - proteinGroup level - how many proteingroups have a specific categorization
DIANN_plot_proteinGroups_upper <- analyze_connected_levels(input_df = DIANN_connected_proteinGroup, connected_levels = "proteinGroup_precursor",count_level = "upper", plot = TRUE, plot_characteristic = "absolute")
Spectronaut_plot_proteinGroups_upper <- analyze_connected_levels(input_df = Spectronaut_connected_proteinGroup, connected_levels = "proteinGroup_precursor", count_level = "upper", plot = TRUE, plot_characteristic = "absolute")
#lower level - precursor level - how many precursor have a specific categorization
DIANN_plot_proteinGroups_lower <- analyze_connected_levels(input_df = DIANN_connected_proteinGroup, connected_levels = "proteinGroup_precursor",count_level = "lower", plot = TRUE, plot_characteristic = "absolute")
Spectronaut_plot_proteinGroups_lower <- analyze_connected_levels(input_df = Spectronaut_connected_proteinGroup, connected_levels = "proteinGroup_precursor", count_level = "lower", plot = TRUE, plot_characteristic = "absolute")
#*Reports*
#ProteinGroup level
DIANN_report_proteinGroups <- analyze_connected_levels(input_df = DIANN_connected_proteinGroup, connected_levels = "proteinGroup_precursor",count_level = "upper", plot = FALSE)
Spectronaut_report_proteinGroups <- analyze_connected_levels(input_df = Spectronaut_connected_proteinGroup, connected_levels = "proteinGroup_precursor",count_level = "lower", plot = FALSE)Example proteinGroup level
kableExtra::kable(DIANN_report_proteinGroups)| Connected_proteinGroups_precursor | absolute_count | relative_count |
|---|---|---|
| common_common | 2 | 20 |
| common_unique | 1 | 10 |
| unique_common | 3 | 30 |
| unique_unique | 4 | 40 |
DIANN_plot_proteinGroups_upper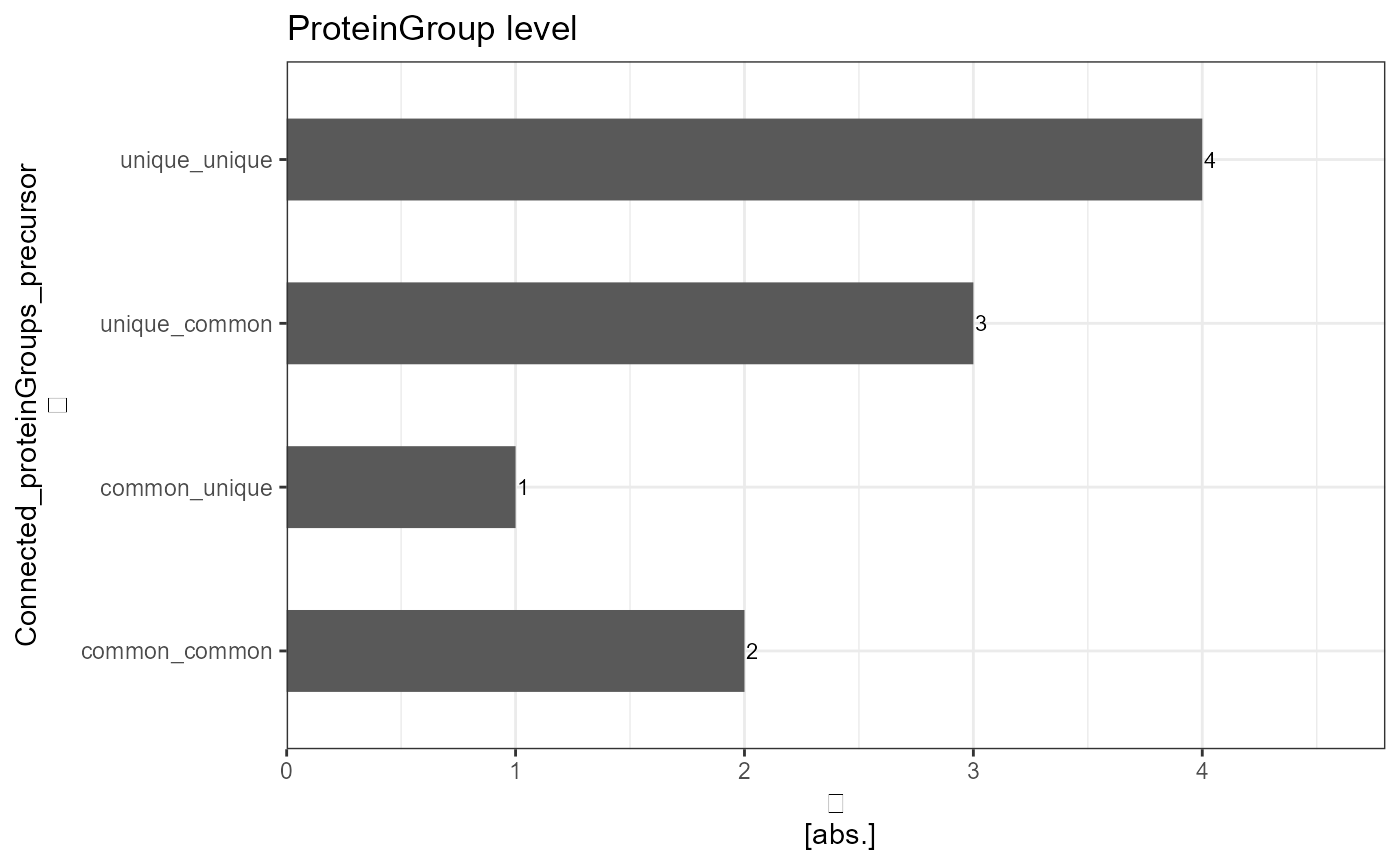
Get software difference for proteinGroup denotations
Filter for potential common precursor and unique proteinGroup connections. It is possible to trace differences in proteinGroup denotations for common precursor.
#with string_analysis = TRUE - if protein denotation is mentioned in both proteinGroups of input_df1/_df2 are filtered out - only distinct protein denotations remain
Difference_proteinGroup <- trace_unique_common_pg(input_df1 = DIANN_connected_proteinGroup, input_df2 = Spectronaut_connected_proteinGroup, analysis_name1 = "DIA-NN", analysis_name2 = "Spectronaut", string_analysis = FALSE)
Difference_proteinGroup_reduced <- trace_unique_common_pg(input_df1 = DIANN_connected_proteinGroup, input_df2 = Spectronaut_connected_proteinGroup, analysis_name1 = "DIA-NN", analysis_name2 = "Spectronaut", string_analysis = TRUE)Results:
First, string_analysis = FALSE is used. ProteinGroups, which share similar proteins, stay in the output. For example, for the common precursor COMMON2 has in Spectronaut a proteinGroup denotation of EXAMPLE2 and in DIA-NN of EXAMPLE1;EXAMPLE2.
| traceR_proteinGroups_DIA-NN | traceR_precursor | traceR_proteinGroups_Spectronaut |
|---|---|---|
| P01764 | AEDTAVYYC(UniMod:4)AK2 | A0A0J9YY99 |
| Q92496 | EGIVEYPR2 | Q02985 |
| EXAMPLE1;EXAMPLE2 | COMMON2 | EXAMPLE2 |
Second, string_analysis = TRUE is applied. ProteiGroups, which have similar proteins, are filtered.
| traceR_proteinGroups_DIA-NN | traceR_precursor | traceR_proteinGroups_Spectronaut |
|---|---|---|
| P01764 | AEDTAVYYC(UniMod:4)AK2 | A0A0J9YY99 |
| Q92496 | EGIVEYPR2 | Q02985 |